






Bio-Share PC&PV | 康日百奥Bioworkshops关于设计一个可比性研究方案的分享
发布时间: Jun 28 , 2023

摘要
在生物制药的开发过程中,生产工艺的变更不可避免。临床试验对样品量的需求,开发者对提高产品收率或纯度的需求以及生产工艺稳健性和可重复性的自身需求,都可能带来生产工艺的变化,而这些变更在当前的生物制药法规中是允许的,但法规同时要求在生产工艺变更前后保持产品的安全性和有效性的可比性至关重要。
可比性研究是生物制药变更评价的基础和成功的关键。可比性研究目的在于通过收集相关技术资料对比分析确认变更有没有对生物制品的质量、安全性和有效性产生不利影响。可比性研究包括工艺可比性、产品质量可比性和稳定性可比性,必要时还应进行非临床及临床研究数据的可比性分析。
设计可比性研究方案
设计可比性研究方案需要考虑变更的类型、预计变更对产品特性造成的影响程度及变更对安全性和有效性潜在影响的评,以及考虑与安全性和有效性相关的产品特性、浓度、质量、纯度或效价等。下面内容将介绍一个科学的可比性研究方案应包含的基本元素。
01. 介绍和背景
介绍部分应包含产品、当前监管状态和生产步骤的概述。背景部分提供了有关为什么提交可比性研究的信息,并详细说明了与生产变更相关的先前监管提交的信息。
02. 变更说明和引入变更的理由
这一部分应包括变更的详细说明,并以表格形式进行展示。包含在工艺中的所有变更并为引入的变更提供充分的理由是至关重要的。在某些情况下,还可能包含分析程序的变更或生产设备的变更。每个变更都应在单独的标题下详细描述。
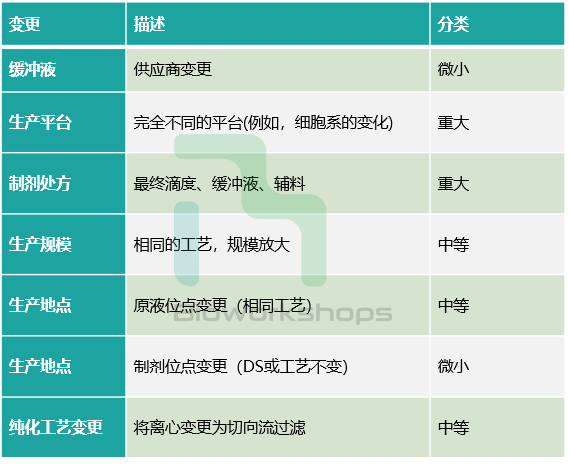
03. 变更的分类
根据《生物制品上市后变更研究技术指导原则》,生产工艺变更可分为微小变更、中度变更和重大变更。微小变更定义为对产品质量没有潜在影响的变更,而中度和重大变更可能对产品质量产生不利影响,可能需要提交新的IND或IND增补。
中度和重大的确定不仅需要产品知识,还需要了解CQAs/CPPs与产品安全性和有效性之间的关系。在CQAs/CPPs与产品质量之间的关系不完全了解的情况下,鼓励使用风险评估原则。
表1. 常规变更的案例及其分类
04. 关键质量属性(CQA)和分析方法
建立产品可比性的核心是申请企业对与产品安全性和生物活性相关的产品特定CQA的了解, CQA构成了可比性研究的主力和主要参考点。关键质量属性及其可接受标准会在生产工艺开发过程中不断完善,并用于设定可比性研究的分析检测对象。
可比性研究中对CQA的分析检测还取决于分析方法的稳健性。分析方法需要处于与可比性研究阶段相对应的控制状态。对于产品早期开发阶段,分析方法的确认即足够。产品后期开发阶段则需要分析方法的验证,否则可比性研究收集的数据集不能代表产品的质量,会影响可比较性研究的结论。
05. 分析检测的操作方法
确定可比性的方法可能涉及对不同生产工艺获得的中间体、原液或制剂材料进行头对头比较。然而有些时候,头对头比较可能不可行,此时也可以将变更后获得的材料的分析数据与变更前获得材料的历史数据进行比较。重要的是确保两种材料使用相同的分析方法进行检测。

06. 建立可比性的可接受标准
可接受标准的选择可能是可比性研究中最具挑战性的一步,建立可比性的可接受标准应在很大程度上依赖于历史产品知识、生产能力和所选分析方法的稳健性。
在提交给监管机构的可比性研究中,生产企业提出了预先确定的可接受标准,该标准应基于历史数据、生产能力和分析可变性来证明是合理的。例如,历史数据在开发的早期阶段可能是有限的,从有限数量的批次中得出预定的可接受标准可能是可以接受的。然而,临床3期研究标准的可接受标准则会基于产品开发生命周期中收集的信息制定,需要额外的说明。
07. 取样计划和统计分析
生产企业应为提议的取样计划提供充分的理由,包括检测的批次数量、用于可比性研究的批次类型以及可比性运行的样品收集方法。用于建立可比性的批次类型应该是可比性研究设计中的一个主要讨论点。这些批次是使用代表大规模生产的工艺(可能在非GMP条件下进行)生产的。
在确定所需样本数量时,选择一种统计方法来建立可比性是一个重要的考虑因素。已经开发了各种统计方法并将其应用于可比性评估。常用的统计方法包括视觉比较、最小值和最大值、置信区间、预测区间、容差区间和等价检验等。
有时,视觉比较(比如并列散点图)可以与统计区间方法(比如置信区间、预测区间等)同时进行。如下图所示:
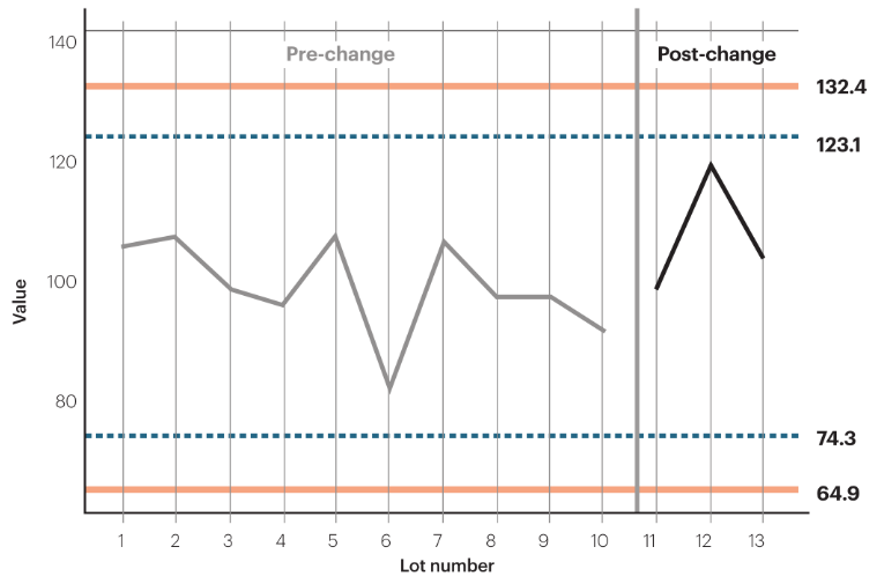
图1. 变更前后批次的视觉比较和统计区间比较。其中(74.3,123.1)为平均值±3SD区间,(64.9,132.4)为95%置信区间。
在有更多批次和更多有关CQA的信息可用的情况下,可以应用严格的统计方法。最严格的统计方法之一是等价检验。合适的等价检验需要适当的样本量和对临床相关性的定量科学理解。
08. 变更后工艺产品的稳定性
可比性的稳定性评估有一个特定的目的,就是必须证明产品在生产后保存在保存场所和临床场所时在一段时间内是稳定的。
稳定性研究也可以实时、加速或在压力条件下进行。加速和压力稳定性研究并不总是可行和/或推荐的。然而,加速稳定性研究(例如,在升高的温度下或在与目标产品相关的其他应力条件下)可以为产品的稳定性提供补充支持证据,并有助于建立温度偏移期间的稳定性曲线。它们通常是确定降解率和/或路径、确定稳定性指标以及提供变更前和变更后产品的直接比较的有用工具。
09. 可比性研究的结论
可比性研究的结论应包含对所收集结果的详细总结,并附有项目的结论。生产企业得出的结论应以简洁全面的形式表达出来。如果数据不完整或收集的信息不支持根据预定的可接受标准建立可比性,则鼓励生产企业加入风险评估部分,以更好地定义产品质量风险并定义后续步骤/未来的计划。
总结
监管机构通常会希望临床3期样品的生产工艺是一个成熟的、明确定义的工艺,并且与最终商业化生产的工艺化一致,不过情况往往并非如此。基于多种原因,生产企业总可能会在临床开发晚期,甚至上市之后发生工艺变更。建立一个科学合理的工艺变更的可比性研究方案至关重要,本篇文章即为目的,总结讲述了一个科学合理的可比性研究方案的主要元素,为充分的、监管认可的可比性研究做好基础。PCPV业务请联系专业的生物药CDMO康日百奥商务小助手:Bioworkshops2019
参考文献
1. Howard L. Levine, Brendan R. Cooney. The development of therapeutic monoclonal antibody products, a comprehensive guide to CMC activities from clone to clinic.
2. ARM & NIMBL, Project A-Gene A case study-based approach to integrating QbD principles in Gene Therapy CMC programs







