






Bio-Share PC&PV | 整合QbD的生物制药制剂开发新路线
发布时间: Apr 30 , 2023

生物制药制剂开发的传统方法
成功制剂开发的目标是保持原液的质量属性(纯度、安全性、含量和效价)稳定,同时确保产品的无菌性。传统液体制剂的开发方法包含以下步骤:
- 利用早期可开发性评估来协助进行分子的选择;
- 通常使用平台处方和平台工艺,结合较少的制剂辅料筛选,以快速进入临床I期和II期;需要进行稳定性评估以支持临床试验中的使用;
- 临床III期处方和工艺的开发需要进行额外的工作,比如评估蛋白浓度、辅料质量、冻融条件、包材相容性,必要时需要微调辅料的浓度范围以确保处方稳健性;
- 申报BLA之前需要进行工艺表征和工艺验证研究(PC/PV),传统方法主要限于3-5批次的商业化规模生产;
与大多数原液生产单元操作不同,制剂工艺极少具有较好的线性缩小模型,加之材料的限制,实际上很难使用大规模的生产工艺对制剂处方和工艺参数进行全面的研究,因此对工艺和产品的理解,以及知识的积累也有限。
整合QbD的制剂开发新路线
质量源于设计(QbD)是一种基于科学和风险的药物开发方法。利用先验知识、平台知识来设定目标产品质量概况(QTPP),基于风险评估来识别关键质量属性(CQA),开发实验室规模的模型并进行多变量实验研究以确定关键工艺参数(CPP)及其设计空间,并制定控制策略。
Genentech曾报道了一种将QbD整合进入生物制药制剂开发的新路线。在新路线中,QbD的元素与工艺表征和工艺验证研究(PC/PV)相结合,从而升级了传统生物制药制剂开发方法。[1]
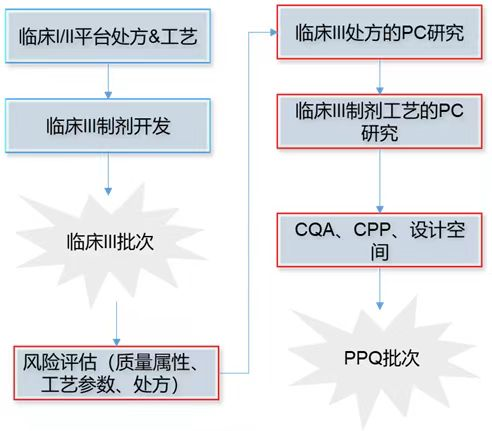
图1. 整合QbD的生物制药制剂开发新路线
CQA:关键质量属性;
CPP:关键工艺参数;
PC:工艺表征;
PPQ:工艺性能确认;
【红色方框是整合的QbD元素】
质量属性的风险评估
制剂质量属性的风险评估类似于原液。通常是评估质量属性对生物活性、PK/PD、安全性和免疫原性的影响程度,以及用于该影响程度评估的信息的置信度,即评估的不确定性。质量属性的总体风险分数为影响程度与不确定性的乘积。当该分数高于某个阈值时,即可确定该质量属性为关键质量属性(CQA),反之则为非关键质量属性。[2]
识别了制剂关键质量属性后,重要的一步是确定关键质量属性与制剂工艺步骤的关系,以确定后续每个制剂单元操作的PC研究的重点。这一步的风险评估可采用因果效应矩阵方法,如下表对假设的单抗制剂进行该风险评估。
表1. 使用风险评估工具评估受制剂单元操作影响关键质量属性
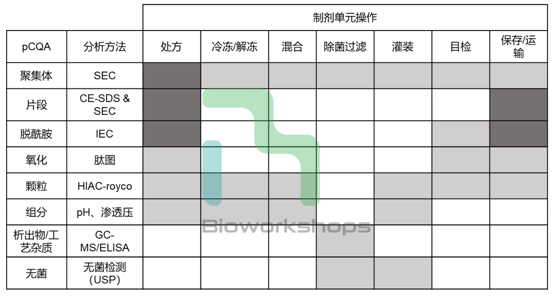
处方组分和制剂工艺参数的风险评估
对处方组分或制剂工艺参数进行风险评估是为了筛选出需要进行PC研究的潜在关键工艺参数。实际上,我们无法对所有的处方组分和工艺参数进行PC的实验性研究,因此需要确定对CQA影响风险较高的处方组分和工艺参数,并对其进行PC研究。
Genentech通过评估处方组分或工艺参数的主效应,以及潜在的交互效应对CQA的影响性来进行评估。最终的风险分数为主效应影响性与交互效应影响性的乘积。风险分数超过一定阈值即确定为潜在关键性工艺参数,需要进入后续的PC研究。制剂工艺参数的风险评估可采用类似方法。
表2. 制剂处方组分的风险评估,使用(主效应影响x交互效应影响)的风险评估工具
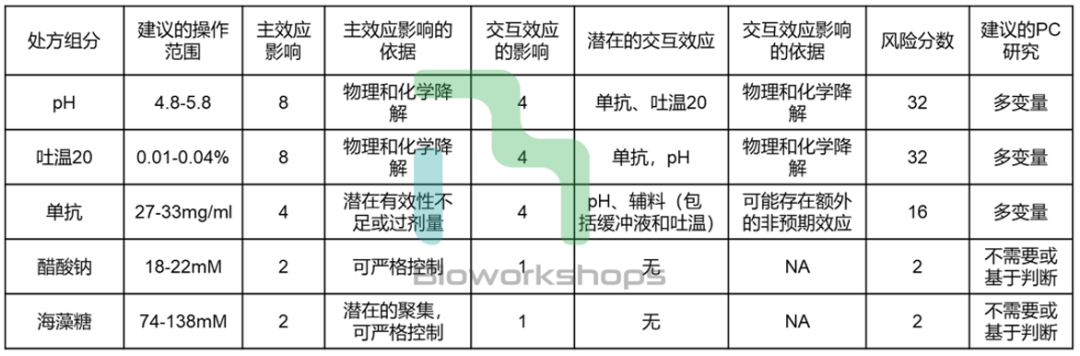
制剂处方的PC研究
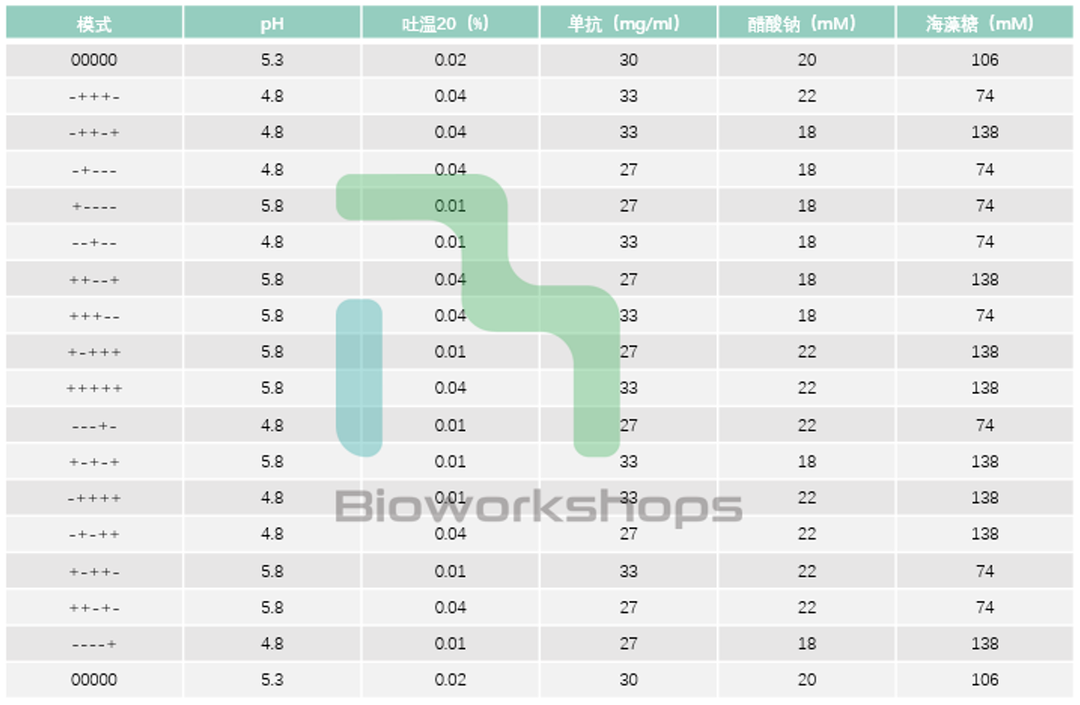
对风险评估的筛选的潜在关键工艺参数可进行实验设计(DoE),比如采用了五因子两水平的部分析因设计,并在三个温度(5°C、25°C 和 40°C)下重复了该设计。
表3. 制剂处方的实验设计(DoE)
实验设计的分析可获得制剂处方中的真正关键工艺参数及其设计空间。以25℃下6个月的实验数据为例进行分析。
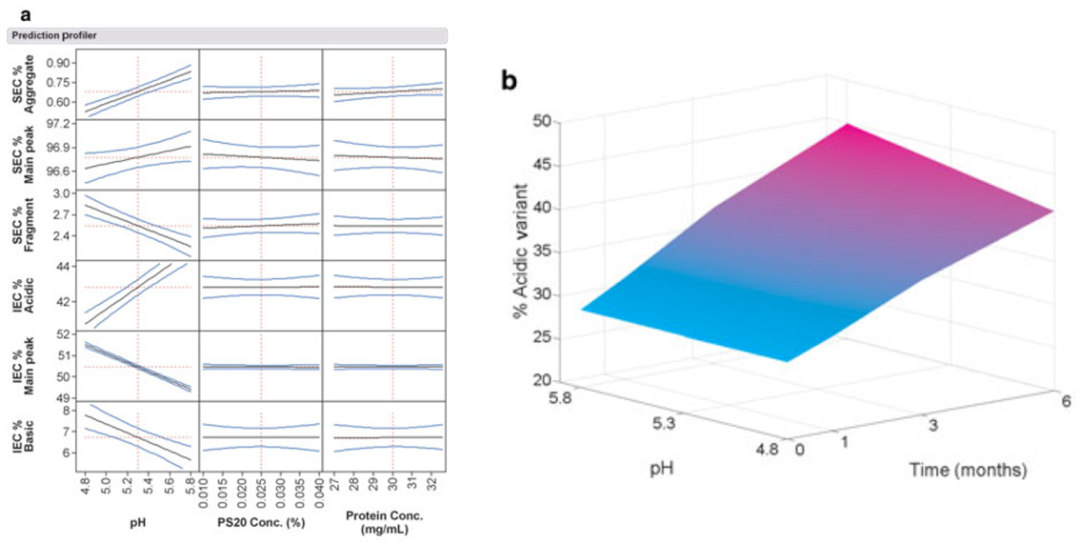
图2. 以25℃下6个月的实验数据进行分析可确定处方组分对CQA的潜在影响,以及设计空间
制剂工艺的PC研究
类似于制剂处方的PC研究,可以对制剂工艺单元操作的风险评估确定的潜在关键工艺参数进行实验室规模的研究。但是需要注意的是,大多数制剂工艺单元操作无法进行精确的线性缩小。因此,在制剂工艺的PC研究中,通常会设计能够代表最差条件的缩小模型,并在该模型上进行PC研究。下面案例展示了按照该思路进行的制剂混合单元操作的PC研究。
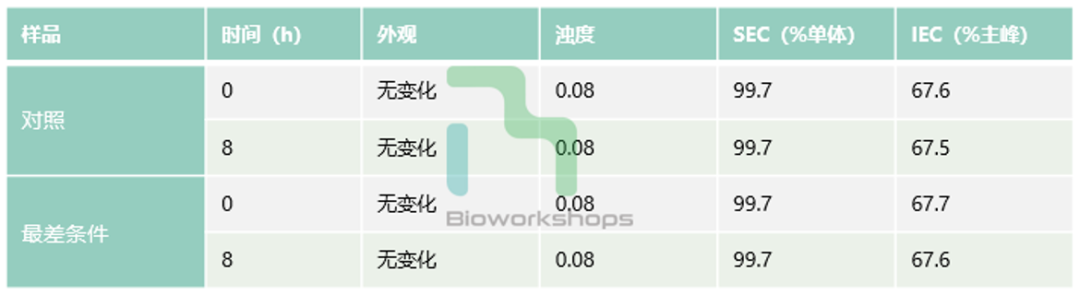
该案例中,单抗原液解冻后转移进600L的配制罐中,并在其中与稀释液进行混合。通过保持重要的几何比例设计了一个缩小模型,其中转速根据单位体积叶轮尖端速度或叶轮功率进行计算,选择计算出的最高速度,同时选择最长混合时间和最高混合温度。该模型则作为最差条件模型进行PC研究。

图3. 混合单元操作的缩小模型设计
最差条件下的研究未观察质量属性的显著变化,因此该单元操作中没有工艺参数确定为关键工艺参数(CPP)。
表4. 混合单元操作的PC研究结果
结论
将QbD元素嵌合进入制剂工艺开发和验证程序中可以带来众多优势,例如增强的产品和工艺知识,支持设计空间的定义以及合理的质量标准设定。
尽管本篇短文只是以制剂处方和制剂工艺的混合单元操作为例进行了阐述,但是这种新路线是可以适用于制剂工艺的所有单元操作的。这种严格的、标准化的流程最终会带来可预测的开发时间表以及审评注册的成功率,对于生物制药质量以及开发公司都会产生有利的影响。
康日百奥Bioworkshops的制剂开发平台可满足不同类型生物制品(如单抗,双抗,单链抗体,纳米抗体和重组蛋白等)的不同处方开发需求。此外,我们还可以提供灌装工艺开发和基于DoE的制剂表征,分子的成药性评估和高浓度制剂的开发服务。欢迎联系专业的生物药CDMO康日百奥Bioworkshops。
参考文献
1. Martin–Moe S, Lim FJ, Wong RL et al (2011) A new roadmap for biopharmaceutical drug product development: integrating development, validation, and quality by design. J Pharm Sci 100:3031–30432. Alavattam Sreedhara, Rita L. Wong, Application of QbD Principles to Late-Stage Formulation Development for Biological Liquid Products







