






Bio-Share PC&PV | 康日百奥浅谈项目晚期质量标准可接受范围的设定
发布时间: May 30 , 2023

前言
药品的质量标准是为了确保获得预期的产品质量而对产品处于适当限制、范围或分布内的物理、化学、生物或微生物性质或特性进行控制的技术要求。根据ICH Q6的定义,药品的质量标准由放行检验项目、分析方法和可接受范围组成,其设定过程包括了放行检测项目的确定、分析方法的建立和验证、可接受范围的确定以及标准物质等方面【1】。本微文的内容讲侧重于介绍项目晚期设定质量标准的可接受范围的方法以及注意事项。
可接受范围的设定方法
可接受范围是指原液、制剂或产品中间体应符合的数值限度、范围或其他合适的测量值。总结法规要求和行业实践,设定项目晚期质量标准的可接受范围的方法主要包括:
➤➤基于患者安全性考虑设定可接受范围
对于患者安全性相关的项目一般都属于此类,比如外源污染物(比如无菌),许多工艺相关杂质(比如宿主细胞DNA残留、宿主细胞蛋白残留、内毒素等)。此类质量标准通常需要不低于药典的要求。
比如无菌,强制性要求,要求无菌检测必须显示细菌/真菌不存在,结果为“无生长”。
比如内毒素,如果注射剂量不超过每名患者每公斤每小时5 EU,则可设定生物药物的注射上限。
对于残留的宿主细胞DNA, 如果注射剂量不超过每剂量10 ng DNA(对于口服疫苗,注射剂量应不超过每剂量100μgDNA),则可以确定生物制药的监管上限。
对于残留的宿主细胞蛋白,生物药物没有官方的监管上限,但是对于重组蛋白和单克隆抗体,可以确定的上限不超过100ppm(即,不超过100 ng/mg蛋白)。
➤➤基于生产工艺性能设定可接受范围
对于纯度、效价等质量属性,通常可基于多批次产品的统计分析来设定可接受范围。对于可选择进行统计分析的批次,监管机构倾向于选择临床试验中使用的批次,不过,生产企业还可以考虑包括(1)生产规模但未用于临床试验的批次,(2)整个临床开发计划中的批次,以及(3)有时可以包含临床前计划或小规模批次开发研究。
可使用的统计方法主要包括
- 最小值/最大值(Min, Max)
通过查看分析中包括的批次的值并选择两个极端值,可以设置基于最小值/最大值的范围。这不是统计方法,并且这种方法不是监管机构推荐使用的方法。
- 参考区间
也称为“ sigma规则”。Sigma(标准偏差)是批次数据在平均值附近的分散(扩散)的度量。平均值±2个标准差覆盖了批次数据的95.5%;平均值±3个标准差涵盖了批次数据的99.7%。使用平均值±3标准差确定的范围似乎是监管机构比较接受统计分析方法。
- 公差区间
这种统计分析与参考区间方法相似,但是它将概率(即置信度%)纳入了范围计算。这种方法认识到范围确定的不确定性随着数据集大小的减小而增加。通常,使用发布的公差区间表来确定一个范围,该范围包含了99%的生产批次数据,并具有95%的置信度。
➤➤基于临床试验研究设定可接受范围
基于工艺性能设定可接受范围,如果选择的批次数量有限,可能会导致统计计算的范围无法涵盖正常生产工艺的变异范围,因此基于临床试验研究结果设定可接受范围可作为一个重要的数据补充。
但通常用于临床II/III期的临床试验样品的批次很少,单纯基于这些批次的数据也无法有效设定可接受范围。临床II期一般会有剂量探索研究,这通常包含高中低剂量的临床试验研究,那么其研究结果可有效用于可接受范围的设定,尤其是产品的有效成分或产品相关杂质的可接受范围。
由于高剂量组受试者会暴露于比正常剂量组更高的杂质水平,在获得高剂量组受试者的安全性结果信息后,可使用高剂量组杂质的暴露制定正常剂量下的杂质可接受范围标准。同理,低剂量组的有效性数据也可以为有效成分含量的可接受范围设定提供合理的依据。Ulli Backofen博士在一次的公开报告中介绍了这种质量标准可接受范围设定的方法案例【2】。
 图1. 临床II期的剂量探索研究数据用于可接受范围设定的案例
图1. 临床II期的剂量探索研究数据用于可接受范围设定的案例
此外,临床试验期间,一些批次的产品会放置一定时间,那么基于这些放置保存后的产品的临床试验数据也可以用于设定可接受范围。比如,有效期末产品临床试验的结果可对药品的货架期标准、有效成分含量的下限、有效性的论证提供合理依据。
可接受范围设定的考虑要点
无论基于何种方法设定质量标准的可接受范围,只要具有合理的数据支撑和科学依据,都是可以接受的。但是在设定质量标准可接受范围时,仍有一些注意事项需要考虑到。
- 统计分析与可接受范围
基于生产工艺性能设定可接受范围需要使用统计分析的方法,这些统计分析方法的一个前提是样本数据分布的正态性。如果通过正态性检验、频数分布观察数据非正态,需要将数据转换为正态分布,或者使用非参数的计算方法。
- 统计分析的批次数量
基于生产工艺性能设定可接受范围,需要考虑样本批次数量。统计分析方法一般建议至少有10批次以上的数据下进行相应的统计分析。如果批次数量有限,那么设定的可接受范围会比较宽。
辉瑞公司的一项研究认为,从统计学分析的角度,25-30批次的数据积累可较好的预测产品工艺能力,进一步积累到85批次可用于确定最终的质量标准可接受范围。
通常,EMA和FDA要求产品上市后累积生产至30批次或累积生产3年后以补充申请方式进行质量标准的更新。
- 分析方法变异与可接受范围设定
采用样本批次数据的统计分析时已经包含了分析方法的变异。如果批次分析数据较多时,把分析方法的变异再添加到可接受范围的设定中,比如可接受范围=多批次数据均值±3SD+分析方法变异,这是不合适的。
如果仅有少量的批次分析数据,或者分析方法的变异与工艺变异相当,那么在一定条件下,并且经监管机构批准,可以将分析方法变异作为一个额外的变异源加以计算。
- 稳定性研究与可接受范围
原液和制剂在储存过程中可能会发生降解,需要对储存过程中产生的明显降解产物予以分析,并纳入质量标准,因此有时需要同时设置货架期标准和放行标准。
基于生产工艺性能设定放行标准的可接受范围,再通过产品在储存期间的稳定性图谱斜率的变化计算货架期标准。Genentech曾在文献中报道了类似的思路【3】。国外某上市疫苗也报道了类似方法设定效价质量标准的可接受范围【4】。
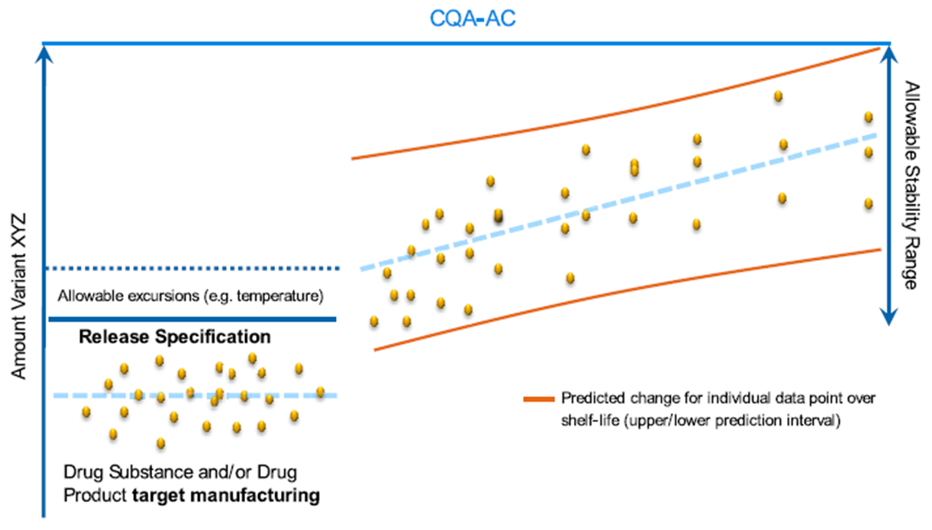 图2. 考虑稳定性研究设定放行标准和货架期标准的可接受范围
图2. 考虑稳定性研究设定放行标准和货架期标准的可接受范围
- 工艺相关杂质的可接受范围
部分工艺相关杂质,比如有机溶剂残留,可以参考ICH Q3C中的可耐受日剂量(TDA)来设定可接受范围,或者参照其他已上市的药品的同类杂质的限度标准,比如吐温80等。
如果没有同类工艺相关杂质的限度标准,可以结合非临床研究和毒理学数据来确定,比如使用杂质安全系数方法(ISF)。
ISF的计算公式如下:
ISF=LD50÷每剂量产品杂质限度
LD50可以通过一些研究资料获得。如果缺少相应的研究中资料,也可以通过毒理学研究获得LD50或者未见不良反应水平(NOAEL)。ISF的设定应考虑足够的大,1000是常用的选项。
总结
项目晚期设定合理的质量质量标准可接受范围会直接影响注册申报,因此需要选择合理的方法以及考虑相应的注意事项。设定质量标准可接受范围的科学依据非常重要。此外,还应在注意质量标准的可接受范围也不是一成不变的,可随着项目的进展,上市后批次数据的积累而不断更新和不断完善。
关于我们
康日百奥生物科技(苏州)有限公司 是一家专业的生物药CDMO,位于苏州工业园区东旺路5号。公司服务范围包括生物药工艺开发、cGMP原液生产、无菌制剂灌装等。团队成员均为经验丰富的生物制药行业资深人士,对生物药CMC领域有着深刻的理解。康日百奥Bioworkshops原液产能13000L,同时拥有包含西林瓶水针,冻干、卡式瓶、预充针、注射笔等的无菌制剂灌装服务,可完全实现从早期临床前样品生产至商业化生产的高效衔接。
康日百奥Bioworkshops 已成功帮助多个合作伙伴的单抗、双抗、多抗、ADC、融合蛋白、细胞因子等项目获得中国、美国、澳大利亚等的临床批件。康日百奥Bioworkshops致力于为全球合作伙伴提供高效、高质量的生物药外包服务解决方案,帮助合作伙伴缩短药物进入临床试验和上市的时间。
公司业务范围:细胞株构建 | 细胞培养 | 纯化工艺开发 | 制剂工艺开发 | 分析方法开发 | 工艺表征工艺验证 | 原液和成品的cGMP生产(200L、500L、2000L) | 无菌灌装(预充针、卡式瓶、注射笔、西林瓶水针、冻干)
参考文献
1.李敏,常卫红,生物制品质量标准研究与建立一般原则的探讨,中国新药杂质,2017年第26卷第16期。
2.Kepert JF, Cromwell M, Engler N, Finkler C, Gellermann G, Gennaro L, et al. Establishing a control system using QbD principles. Biologicals 2016;44(5):319e31.
3.Dr.UlliBackofen& Dr.Rico Lippmann Justification of specification &life-cycle managementof relevant analytical methods CMC Strategy Forum | China 2021.
4.CAPEN R,SHANK-RETZLAFF M,SINGS H,et al. Establishing potency specifications for antigen vaccines[J]. Bio Process Int,2007,5( 5) : 30 - 42.







